
In April 2026, the Parkinson’s Foundation hosted an expert briefing titled “Inside the Science: Parkinson’s Research Today.” Laurie Sanders, PhD, associate professor of neurology at Duke University, shared why she and many of her colleagues are increasingly optimistic about the direction of Parkinson’s disease research. Dr. Sanders focused on three key research areas — alpha-synuclein, mitochondrial function, and neuroinflammation — and explained how these pathways are now understood to be interconnected, opening new doors for disease-modifying therapies. This was a technical webinar. If this talk taught me anything, it’s that Parkinson’s disease is far more complex than most of us realize. I found myself looking up terms throughout — so consider this my best attempt at summarizing the science.
Key Takeaways
The science of Parkinson’s disease is advancing at a remarkable pace. Parkinson’s is now understood as a whole-body, multisystem disease — not just a motor disorder. Researchers have a much deeper understanding of the genetic, molecular, and cellular drivers of PD than ever before, and there are currently over 200 active or soon-to-open clinical trials. There is a real reason for optimism.
Alpha-synuclein is a central but complex target in Parkinson’s disease. Alpha-synuclein is a normal protein in the brain that helps with neurotransmitter release and communication between neurons. In Parkinson’s, it misfolds, clumps, and spreads between cells, forming Lewy bodies and driving cellular dysfunction. Researchers are working to selectively target the misfolded forms without disrupting the protein’s normal function. There are nearly 100 known conformations (strains) of synuclein, and identifying which are most toxic is a key current focus.
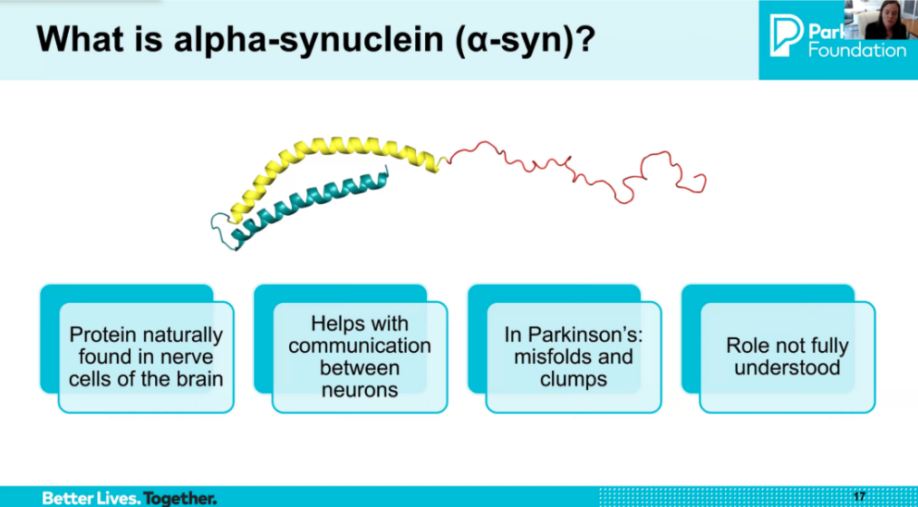
Image source: slide presentation
Mitochondria are far more than the powerhouses of the cell — and their dysfunction plays a central role in PD. Mitochondria manage energy production, DNA repair, cell death signaling, and many other critical functions. In Parkinson’s, mitochondrial dysfunction has been observed in brain tissue, muscle, and blood. Mutations in some genes are linked to mitochondrial dysfunction and PD. Environmental toxins such as paraquat, rotenone, and MPTP reproduce features of PD in animal models through the same mitochondrial pathway.
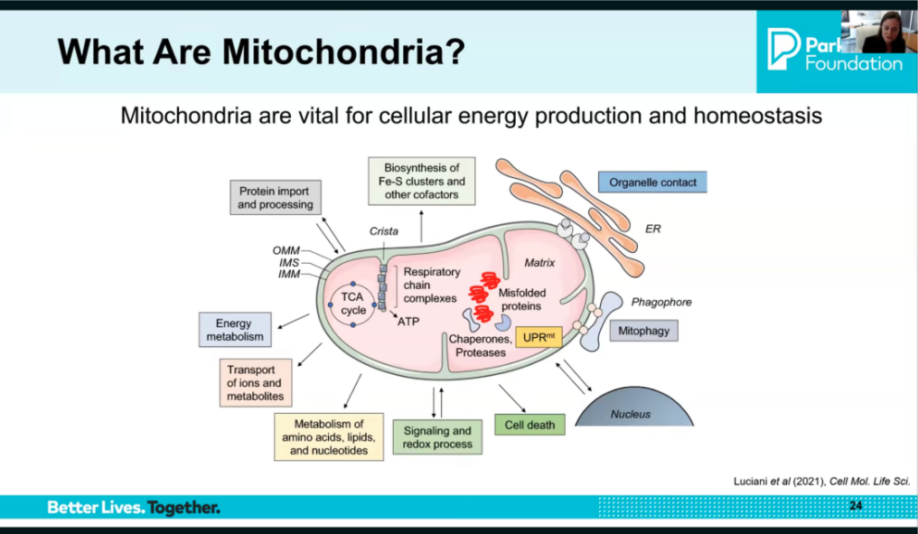
Image source: slide from presentation
A new blood-based biomarker for mitochondrial dysfunction may transform how clinical trials are designed. Dr. Sanders’ lab at Duke has developed a PCR-based blood test called MtDNA-DX that measures mitochondrial DNA damage as a biomarker of early Parkinson’s disease. This test can go from blood sample to result in a single day and is being moved toward use in clinical trials. The goal is precision medicine — matching people with PD to the right therapy based on their specific underlying biology, much like biomarker-driven approaches already used in cancer treatment.
Neuroinflammation is a key — and interconnected — driver of Parkinson’s disease. Brain immune cells called microglia normally help clean up damage, but in PD they can become chronically activated, releasing inflammatory substances that damage neurons in a vicious cycle. Synuclein aggregates and dying neurons trigger microglia activation; a leaky blood-brain barrier allows peripheral immune cells into the brain; and gut inflammation may also play a role through the gut-brain axis. PET imaging using a marker called TSPO can now measure this neuroinflammation in living people.
Alpha-synuclein, mitochondrial dysfunction, and neuroinflammation are not separate problems — they are deeply interconnected. These three pathways drive and amplify each other, and PD is now understood as arising from the interaction between them rather than from any one pathway in isolation. This interconnected understanding is why researchers are increasingly interested in combination therapies that target multiple pathways simultaneously, and why past single-pathway clinical trials may not have succeeded.
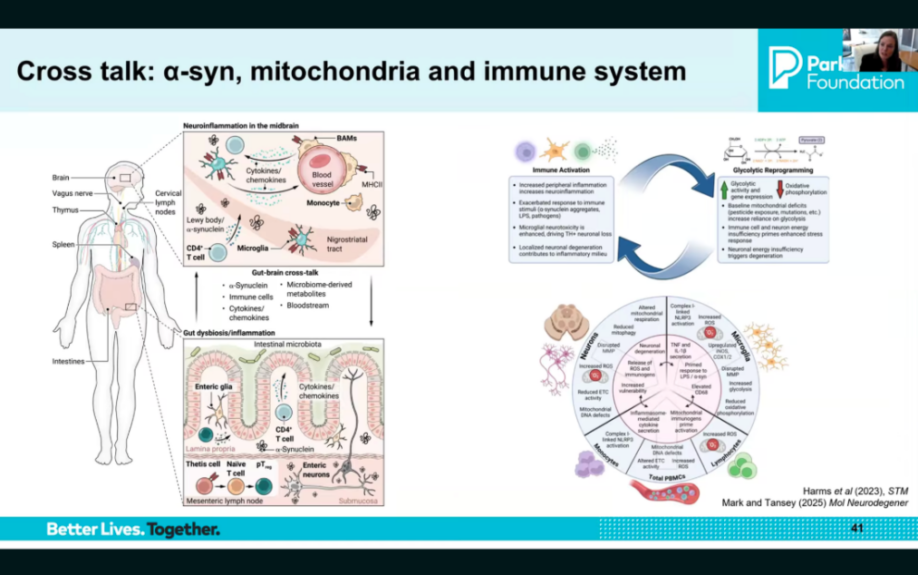
Image source: slide from presentation
The field is moving toward precision medicine and disease-modifying therapies. Rather than treating symptoms alone, the goal is now to identify the specific biological drivers in each person with PD and match them to targeted therapies. Biomarkers — including the synuclein seeding amplification assay (SAA), neurofilament light (NfL), and the mitochondrial DNA damage assay — are making this possible. Combination therapies that address multiple pathways at once are on the horizon.
Resources
- Webinar Recording — Parkinson’s Foundation YouTube
- “Gut-Brain & Parkinson’s Disease” – Notes from Stanford Seminar, January 10, 2026
Continue reading for detailed notes,
-Elizabeth
Expert Briefing: Inside the Science: Parkinson’s Research Today
Speaker: Laurie Sanders, PhD, associate professor of neurology, Duke University in Durham, North Carolina
Webinar Host: Parkinson’s Foundation
Webinar Date: April 8, 2026
Summary by: Elizabeth Wong, Stanford Parkinson’s Community Outreach
The Big Picture: Why Now Is an Exciting Time in Parkinson’s Research
Parkinson’s disease is the most common movement neurodegenerative disorder, with over 11 million cases globally and over 1 million in the US alone. Projections estimate this will exceed 25 million cases by 2050. In 2024, PD cost the US $82.2 billion. While there is no cure yet, the pace of progress is accelerating rapidly.
The evolution of scientific understanding of PD started from James Parkinson’s original essays describing motor symptoms, to the discovery of dopamine loss in the substantia nigra, to the development of levodopa therapy in 1961, to today’s recognition of PD as a whole-body, multisystem disease with both motor and non-motor features, complex genetic underpinnings, and multiple interconnected cellular pathways.
Key drivers of recent progress include:
- Discovery of over 90 genetic loci (location on a chromosome) associated with PD risk, providing a window into underlying cellular dysfunction
- Advances in imaging including PET ligands and high-resolution MRI
- Development of biomarkers including the alpha-synuclein seeding amplification assay (SAA), neurofilament light (NfL), and inflammatory markers
- New molecular tools including single-cell RNA sequencing, CRISPR screens, cryo-EM structures, iPSC-derived neurons and organoids
- Global collaboration and patient participation in research
- A rapidly growing clinical trial pipeline with over 200 active or soon-to-open trials
Alpha-Synuclein: A Central but Complex Target
Alpha-synuclein is a normal protein found in all brain cells. It plays an important physiological role in the presynaptic part of neurons, helping with neurotransmitter release (including dopamine), vesicular trafficking, and communication between neurons. Because it has this normal function, selectively targeting only the pathological forms — without disrupting the healthy ones — is a key challenge.
In Parkinson’s disease, alpha-synuclein begins to misfold and clump together, forming oligomers and fibrils that aggregate into Lewy bodies — a hallmark of PD pathology. There is now strong evidence that synuclein can propagate between cells in a prion-like manner, and its interaction with lipid membranes and organelles contributes to mitochondrial dysfunction and inflammation. There are nearly 100 known conformations (strains) of synuclein, and identifying which are most toxic — and therefore the best to target — is a major current focus of the field.
Point mutations and multiplications in the synuclein gene cause familial PD. Genetic modifiers such as GBA and LRRK2 influence synuclein biology and risk. Synuclein pathology is correlated with cognition and disease progression.
Synuclein is not only found in the brain — it is also present in the olfactory system and the gut, fueling research into whether PD originates in the gut and spreads to the brain, or vice versa. The Gut Brain Parkinson’s Consortium was launched by Dr. Sanders’ team to study GI symptoms and the gut-brain connection in PD over time.
Current therapeutic approaches targeting synuclein include reducing production, blocking aggregation, clearing aggregates through immunotherapy, boosting glucocerebrosidase (GBA) activity, and enhancing protein clearance pathways.
Mitochondrial Dysfunction: Much More Than Energy
Mitochondria are double-membrane organelles found in nearly every cell, containing their own DNA and performing many functions beyond energy production: protein import and processing, biosynthesis of iron-sulfur clusters, organelle signaling (including with the endoplasmic reticulum and nucleus), mitophagy (selective removal of dysfunctional mitochondria), redox signaling, and amino acid and lipid metabolism. Critically, mitochondria also serve as key regulators of whether a neuron lives or dies.
In Parkinson’s disease, mitochondrial dysfunction — including a specific defect in mitochondrial Complex I of the respiratory chain — has been observed in brain tissue as well as systemically in muscle and blood. Mitochondrial Complex I is the first and largest protein “machine” located in a mitochondria and acts as the primary gatekeeper to make energy. Environmental toxins such as paraquat, rotenone, and MPTP reproduce features of PD in animal models through this same Complex I pathway. Mutations in DJ1, PINK1, Parkin, LRRK2, and POLG are all linked to mitochondrial dysfunction and PD. Impaired mitophagy — the normal process by which dysfunctional mitochondria are cleared — is thought to contribute to an accumulation of damaged mitochondria in PD.
Dopamine neurons are especially vulnerable to mitochondrial dysfunction because of their uniquely long axons, which travel from the substantia nigra all the way to the striatum, requiring mitochondria to function well at great distances from the cell body.
Dr. Sanders’ lab has developed a blood-based biomarker of mitochondrial DNA damage called MtDNA-DX. This PCR-based assay measures whether mitochondrial DNA damage is out of homeostasis — either due to increased damage or impaired repair — and can go from blood sample to result in a single day. The goal is to use this biomarker to identify people with PD who have mitochondrial dysfunction as a primary driver, so they can be matched to mitochondrial-targeted therapies in clinical trials. This represents a precision medicine approach to PD, similar to biomarker-driven treatment strategies long used in oncology.
Therapeutic strategies targeting mitochondria include mitophagy enhancers, metabolic enhancers through the Nicotinamide Adenine Dinucleotide (NAD+) pathway, and gene therapy approaches to rescue mitochondrial function.
Neuroinflammation: Friend, Foe, or Both
The brain’s immune cells — microglia and astrocytes — normally respond to damage by releasing protective chemicals and clearing debris. In Parkinson’s disease, this response can become chronic and dysregulated, contributing to neuronal damage in a self-amplifying cycle. Synuclein aggregates and dying neurons trigger microglia activation; activated microglia release cytokines and other inflammatory substances; and the resulting inflammation further damages neurons and activates more microglia.
A leaky blood-brain barrier allows peripheral immune cells — normally excluded from the brain — to enter, adding to the inflammatory burden. Gut inflammation and dysbiosis (too few beneficial bacteria, too many harmful ones, or low diversity of the microbiome) may also contribute through the gut-brain axis, potentially via the vagus nerve. Normal aging further compounds this by impairing immune cell function through immunosenescence.
PET imaging using a marker called TSPO now makes it possible to measure neuroinflammation in the living brain, providing a new tool for both research and clinical trials.
Key open questions in the inflammation field include understanding whether immune activation is a cause or consequence of synuclein pathology and mitochondrial dysfunction, how immune signatures vary across disease stages, and how to selectively dampen harmful inflammation without impairing the immune system’s protective functions.
Therapeutic strategies include anti-inflammatory agents, microglia and innate immune modulators, adaptive immune strategies, peripheral immune targeting, gut-targeted therapies, and vagus nerve stimulation.
How These Pathways Are Interconnected
Alpha-synuclein, mitochondrial dysfunction, and neuroinflammation are no longer viewed as separate problems — they interact with and amplify each other. Synuclein aggregates can trigger mitochondrial dysfunction and immune activation; mitochondrial dysfunction can contribute to immune exhaustion and further synuclein misfolding; and chronic inflammation can worsen both. This interconnected understanding has two major implications: it may help explain why single-pathway clinical trials have not yet succeeded, and it points toward combination therapies targeting multiple pathways simultaneously as a more promising approach.
Where the Field Is Headed
Some key directions for the future of PD research and treatment are:
- Earlier detection: The synuclein seeding amplification assay (SAA) and other biomarkers are enabling diagnosis at earlier biological stages of disease
- Precision medicine: Biomarkers will allow researchers to match specific therapies to specific patients based on their underlying biology
- Combination therapies: Targeting multiple interconnected pathways simultaneously
- Disease-modifying therapies: Moving beyond symptom management toward halting or slowing disease progression
- Gut-brain research: Better understanding the role of the gut and peripheral nervous system in PD onset and progression
Question and Answers
Q: No two people with Parkinson’s have the same symptoms. How does that impact your research, and do you think there is a biological basis for this heterogeneity?
A: The heterogeneity in Parkinson’s disease — both in symptoms and in the underlying biology — is something I have thought deeply about since my postdoctoral training, when I began meeting directly with people with PD to understand their experiences. My lab takes a patient-first approach: starting with human studies and patient-derived material to understand what is actually happening across a wide range of individuals, then modeling specific aspects in the lab, and then bringing findings back to human cells to validate them. Current animal and cell models do not adequately capture the complexity and heterogeneity of human PD, and that has been a significant barrier to progress.
Q: Why have so many clinical trials for Parkinson’s disease failed to reach their endpoints?
A: I think there are two key reasons. First, existing models have been inadequate in capturing the full complexity of PD pathology, meaning that findings in preclinical studies have not always translated to humans. Second, trials have tended to target one pathway at a time, whereas the disease involves multiple interconnected pathways working together. A multi-pronged approach targeting synuclein, mitochondria, and the immune system simultaneously may be needed to truly move the needle.
Q: Is alpha-synuclein aggregation the cause of Parkinson’s disease, or could it be a consequence of other upstream dysfunction?
A: This is controversial. Patients with the LRRK2 mutation can have Parkinson’s disease without synuclein aggregation, suggesting that synuclein pathology is not the only route to PD. That said, synuclein pathology is known to occur very early — potentially in the gut decades before clinical symptoms — and clearly drives disease in many people. My view is that different people may reach the same clinical endpoint through different initial triggers, and that understanding those triggers will be critical for developing the right disease-modifying therapies for the right patients.
Q: What role do pesticides and environmental toxins play in Parkinson’s disease?
A: Many environmental toxins associated with increased PD risk — including paraquat, rotenone, and MPTP — share a common mechanism: they disrupt complex one of the mitochondrial respiratory chain. Rather than focusing on specific exposures, I think it is more productive to understand the pathway dysfunction those exposures trigger — particularly mitochondrial dysfunction — and then identify how to block or reverse that dysfunction. This approach could potentially benefit people across different causes of PD, whether genetic, environmental, or idiopathic.